# (PART) Case studies {-}
# Human PBMCs (10X Genomics)
## Introduction
This performs an analysis of the public PBMC ID dataset generated by 10X Genomics [@zheng2017massively],
starting from the filtered count matrix.
## Data loading
```r
library(TENxPBMCData)
all.sce <- list(
pbmc3k=TENxPBMCData('pbmc3k'),
pbmc4k=TENxPBMCData('pbmc4k'),
pbmc8k=TENxPBMCData('pbmc8k')
)
```
## Quality control
```r
unfiltered <- all.sce
```
Cell calling implicitly serves as a QC step to remove libraries with low total counts and number of detected genes.
Thus, we will only filter on the mitochondrial proportion.
```r
library(scater)
stats <- high.mito <- list()
for (n in names(all.sce)) {
current <- all.sce[[n]]
is.mito <- grep("MT", rowData(current)$Symbol_TENx)
stats[[n]] <- perCellQCMetrics(current, subsets=list(Mito=is.mito))
high.mito[[n]] <- isOutlier(stats[[n]]$subsets_Mito_percent, type="higher")
all.sce[[n]] <- current[,!high.mito[[n]]]
}
```
```r
qcplots <- list()
for (n in names(all.sce)) {
current <- unfiltered[[n]]
colData(current) <- cbind(colData(current), stats[[n]])
current$discard <- high.mito[[n]]
qcplots[[n]] <- plotColData(current, x="sum", y="subsets_Mito_percent",
colour_by="discard") + scale_x_log10()
}
do.call(gridExtra::grid.arrange, c(qcplots, ncol=3))
```
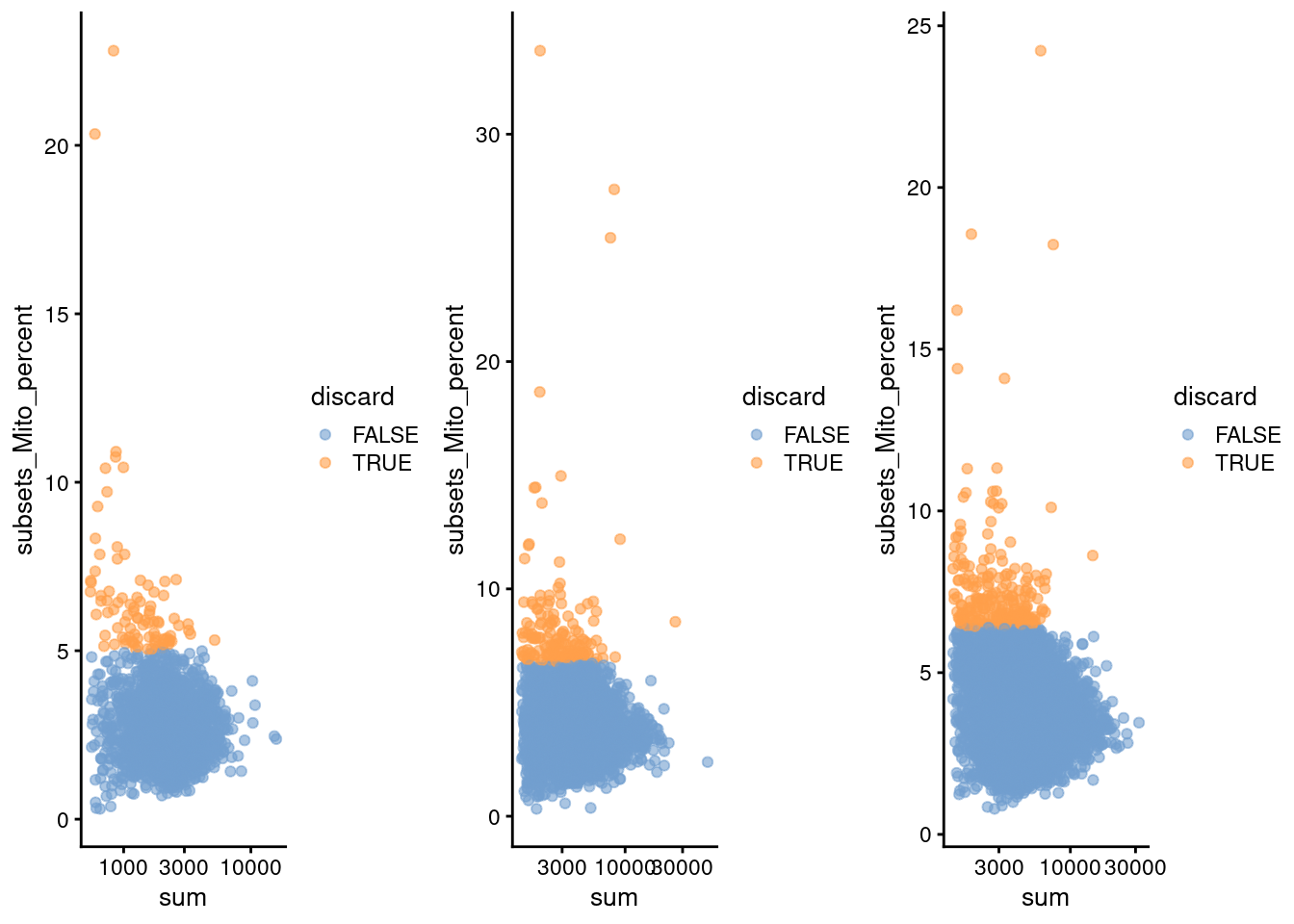
(\#fig:unref-pbmc-filtered-var)Percentage of mitochondrial reads in each cell in each of the 10X PBMC datasets, compared to the total count. Each point represents a cell and is colored according to whether that cell was discarded.
```r
lapply(high.mito, summary)
```
```
## $pbmc3k
## Mode FALSE TRUE
## logical 2609 91
##
## $pbmc4k
## Mode FALSE TRUE
## logical 4182 158
##
## $pbmc8k
## Mode FALSE TRUE
## logical 8157 224
```
## Normalization
We perform library size normalization, simply for convenience when dealing with file-backed matrices.
```r
all.sce <- lapply(all.sce, logNormCounts)
```
```r
lapply(all.sce, function(x) summary(sizeFactors(x)))
```
```
## $pbmc3k
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.234 0.748 0.926 1.000 1.157 6.604
##
## $pbmc4k
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.315 0.711 0.890 1.000 1.127 11.027
##
## $pbmc8k
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.296 0.704 0.877 1.000 1.118 6.794
```
## Variance modelling
```r
library(scran)
all.dec <- lapply(all.sce, modelGeneVar)
all.hvgs <- lapply(all.dec, getTopHVGs, prop=0.1)
```
```r
par(mfrow=c(1,3))
for (n in names(all.dec)) {
curdec <- all.dec[[n]]
plot(curdec$mean, curdec$total, pch=16, cex=0.5, main=n,
xlab="Mean of log-expression", ylab="Variance of log-expression")
curfit <- metadata(curdec)
curve(curfit$trend(x), col='dodgerblue', add=TRUE, lwd=2)
}
```
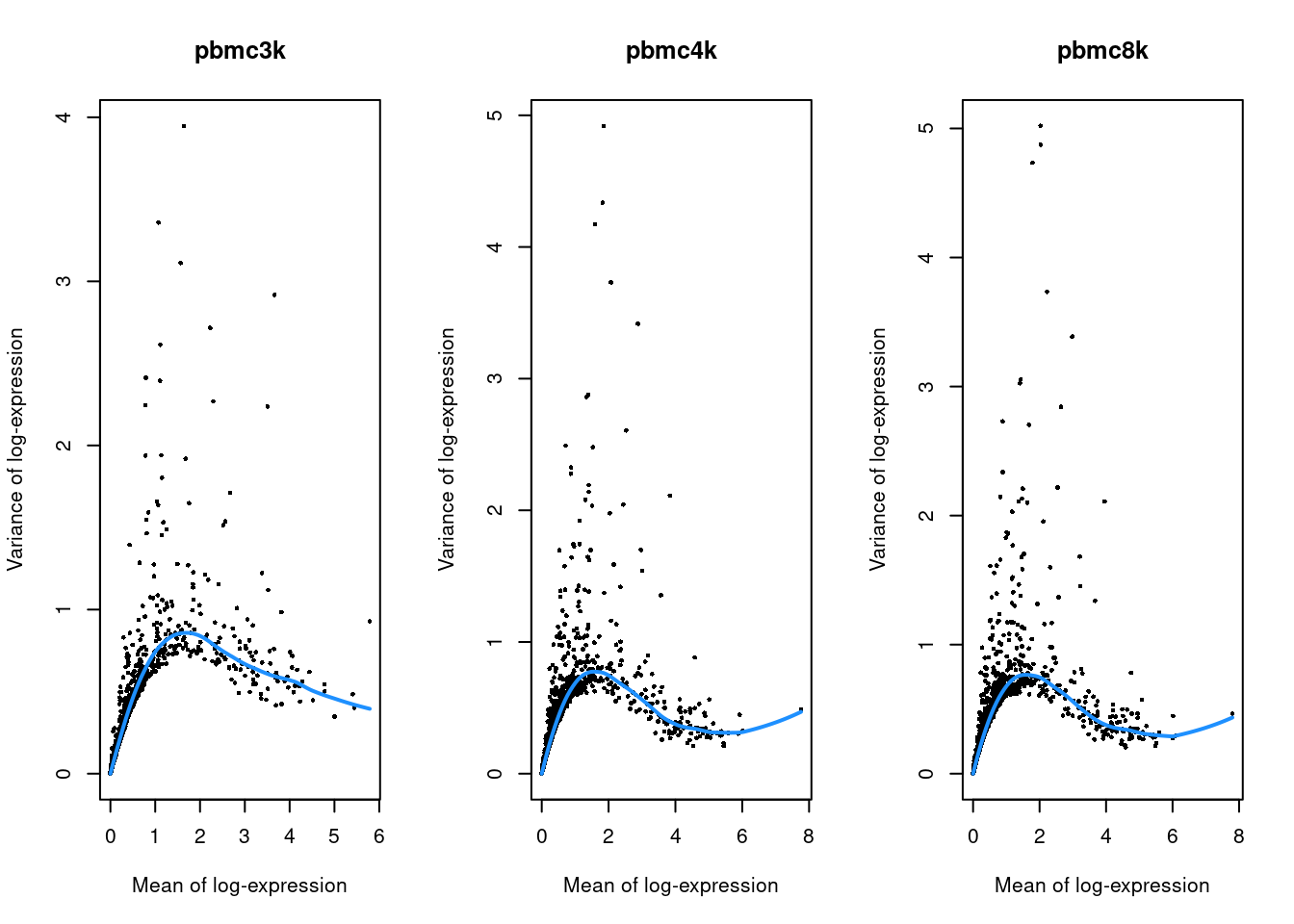
(\#fig:unref-filtered-pbmc-variance)Per-gene variance as a function of the mean for the log-expression values in each PBMC dataset. Each point represents a gene (black) with the mean-variance trend (blue) fitted to the variances.
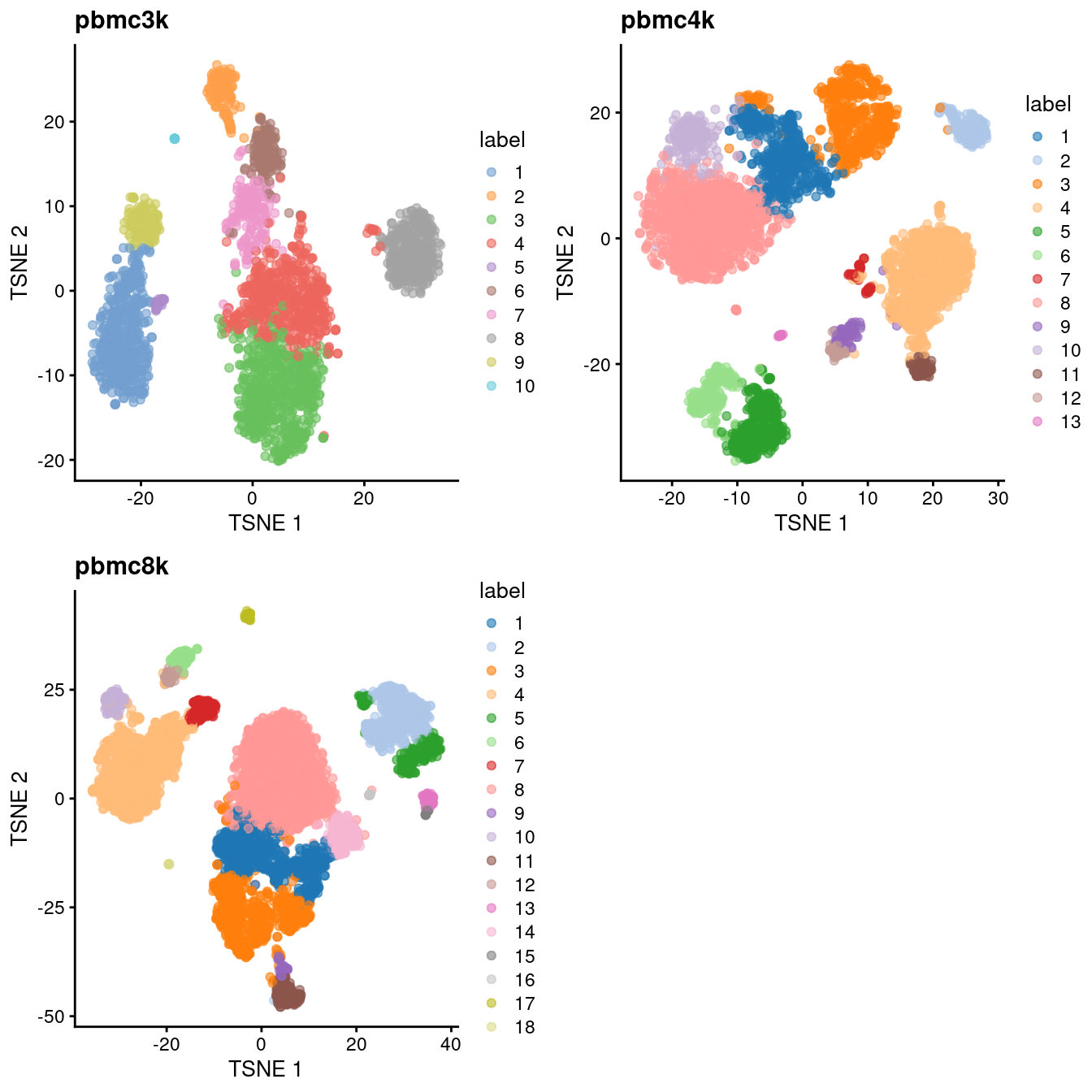
(\#fig:unref-filtered-pbmc-tsne)Obligatory $t$-SNE plots of each PBMC dataset, where each point represents a cell in the corresponding dataset and is colored according to the assigned cluster.
## Data integration
With the per-dataset analyses out of the way, we will now repeat the analysis after merging together the three batches.
```r
# Intersecting the common genes.
universe <- Reduce(intersect, lapply(all.sce, rownames))
all.sce2 <- lapply(all.sce, "[", i=universe,)
all.dec2 <- lapply(all.dec, "[", i=universe,)
# Renormalizing to adjust for differences in depth.
library(batchelor)
normed.sce <- do.call(multiBatchNorm, all.sce2)
# Identifying a set of HVGs using stats from all batches.
combined.dec <- do.call(combineVar, all.dec2)
combined.hvg <- getTopHVGs(combined.dec, n=5000)
set.seed(1000101)
merged.pbmc <- do.call(fastMNN, c(normed.sce,
list(subset.row=combined.hvg, BSPARAM=RandomParam())))
```
We use the percentage of lost variance as a diagnostic measure.
```r
metadata(merged.pbmc)$merge.info$lost.var
```
```
## pbmc3k pbmc4k pbmc8k
## [1,] 7.003e-03 3.126e-03 0.000000
## [2,] 7.137e-05 5.125e-05 0.003003
```
We proceed to clustering:
```r
g <- buildSNNGraph(merged.pbmc, use.dimred="corrected")
colLabels(merged.pbmc) <- factor(igraph::cluster_louvain(g)$membership)
table(colLabels(merged.pbmc), merged.pbmc$batch)
```
```
##
## pbmc3k pbmc4k pbmc8k
## 1 508 395 810
## 2 327 588 1125
## 3 185 122 218
## 4 150 180 294
## 5 175 340 581
## 6 292 542 1014
## 7 347 638 1202
## 8 436 760 1558
## 9 17 27 111
## 10 114 388 832
## 11 33 109 179
## 12 11 54 160
## 13 11 3 9
## 14 3 36 64
```
And visualization:
```r
set.seed(10101010)
merged.pbmc <- runTSNE(merged.pbmc, dimred="corrected")
gridExtra::grid.arrange(
plotTSNE(merged.pbmc, colour_by="label", text_by="label", text_colour="red"),
plotTSNE(merged.pbmc, colour_by="batch")
)
```
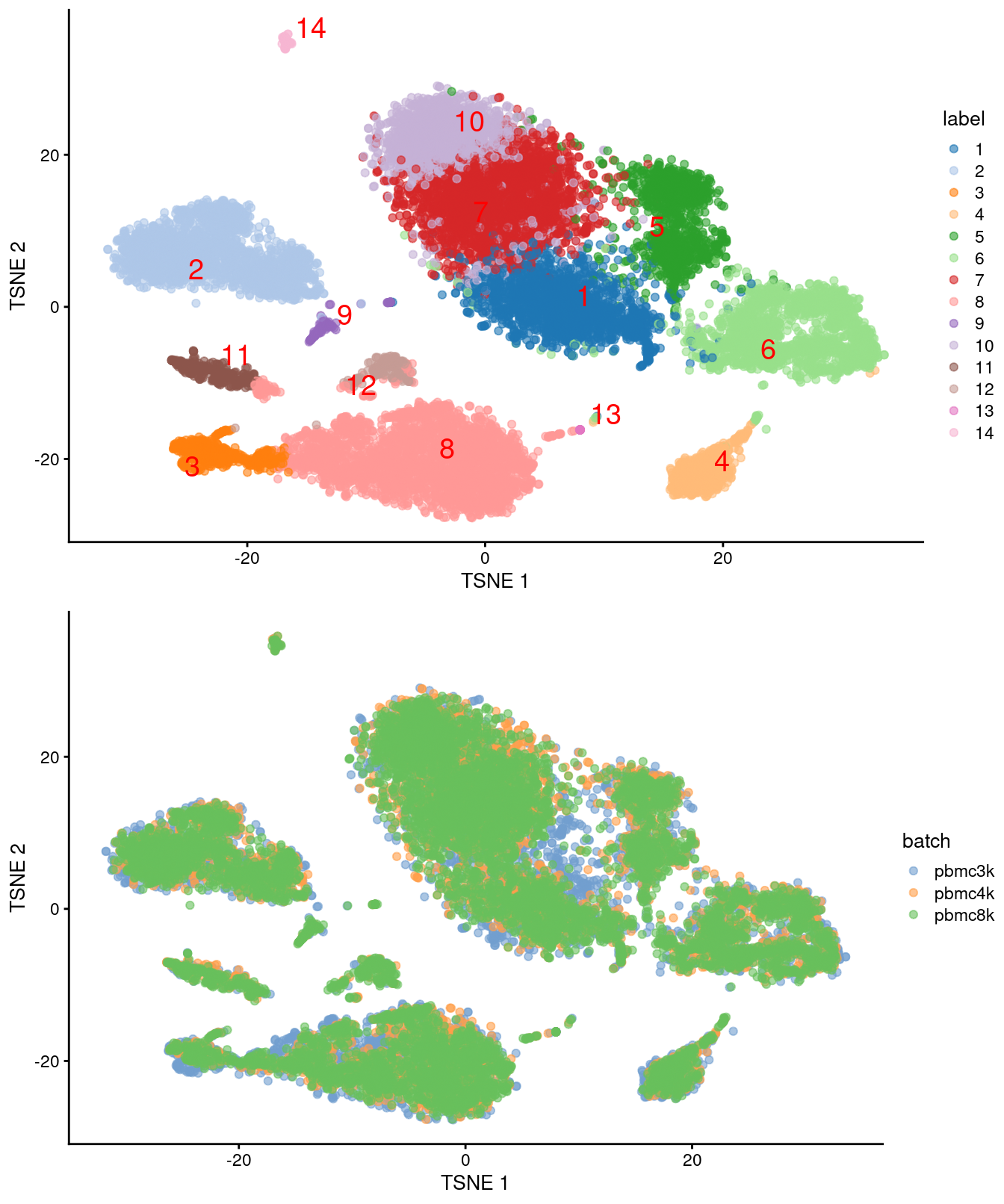
(\#fig:unref-filtered-pbmc-merged-tsne)Obligatory $t$-SNE plots for the merged PBMC datasets, where each point represents a cell and is colored by cluster (top) or batch (bottom).